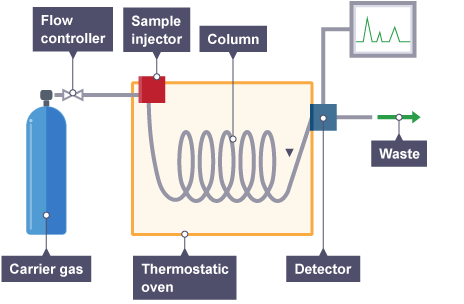
PROBLEMS IN GAS CHROMATOGRAPHY
TROUBLESHOOTING:
EVALUATING THE PROBLEM:
The first step in any troubleshooting effort is to step back and evaluate the situation. Rushing to solve the problem often results in a critical piece of important information being overlooked or neglected. In addition to the problem, look for any other changes or differences in the chromatogram. Many problems are accompanied by other symptoms. Retention time shifts, altered baseline noise or drift, or peak shape changes are only a few of the other clues that often point to or narrow the list of possible causes. Finally, make note of any changes or differences involving the sample. Solvents, vials, pipettes, storage conditions, sample age, extraction or preparation techniques, or any other factor influencing the sample environment can be responsible.
SIMPLE CHECKS AND OBSERVATIONS:
A surprising number of problems involve fairly simple and often overlooked components of the GC system or analysis. Many of these items are transparent in the daily operation of the GC and are often taken for granted (set it and forget it). The areas and items to check include:
1. Gases - pressures, carrier gas average linear velocity, and flow rates (detector, split vent, septum purge).
2. Temperatures - column, injector, detector and transfer lines.
3. System parameters - purge activation times, detector attenuation and range, mass ranges, etc.
4. Gas lines and traps - cleanliness, leaks, expiration.
5. Injector consumables - septa, liners, O-rings and ferrules.
6. Sample integrity - concentration, degradation, solvent, storage.
7. Syringes - handling technique, leaks, needle sharpness, cleanliness.
8. Data system - settings and connections.
GHOST PEAKS AND CARRYOVER:
System contamination is responsible for most ghost peaks or carryover problems. If the extra ghost peaks are similar in width to the sample peaks (with similar retention times), the contaminants were most likely introduced into the column at the same time as the sample. The extra compounds may be present in the injector (i.e., contamination) or in the sample itself. Impurities in solvents, vials, caps and syringes are only some of the possible sources. Injecting sample and solvent blanks may help to find possible sources of the contaminants. If the ghost peaks are much broader than the sample peaks, the contaminants were most likely already in the column when the injection was made. These compounds were still in the column when a previous GC run was terminated. They elute during a later run and are often very broad. Sometimes numerous ghost peaks from multiple injections overlap and elute as a hump or blob. This often takes on the appearance of baseline drift or wander.
Increasing the final temperature or time in the temperature program is one method to minimize or eliminate a ghost peak problem. Alternatively, a short bake-out after each run or series of runs may remove the highly retained compounds from the column before they cause a problem. Performing a condensation test is a good method to determine whether a contaminated injector is the source of the carryover or ghost peaks.
Column Degradation and Contamination
COLUMN BREAKAGE:
Fused silica columns break wherever there is a weak point in the polyimide coating. The polyimide coating protects the fragile fused silica tubing. The continuous heating and cooling of the oven, vibrations caused by the oven fan and being wound on a circular cage all place stress on the tubing.
Eventually breakage occurs at a weak point. Weak spots are created when the polyimide coating is scratched or abraded. This usually occurs when a sharp point or edge is dragged over the tubing. Column hangers and tags, metal edges in the GC oven, column cutters and miscellaneous items on the lab bench are just some of the common sources of sharp edges or points.
It is rare for a column to spontaneously break. Column manufacturing practices tend to expose any weak tubing and eliminate it from use in finished columns. Larger diameter columns are more prone to breakage. This means that greater care and prevention against breakage must be taken with 0.45-0.53 mm I.D. tubing than with 0.18-0.32 mm I.D. tubing.
A broken column is not always fatal. If a broken column was maintained at a high temperature either continuously or with multiple temperature program runs, damage to the column is very likely. The back half of the broken column has been exposed to oxygen at elevated temperatures which rapidly damages the stationary phase.
The front half is fine since carrier gas flowed through this length of column. If a broken column has not been heated or only exposed to high temperatures or oxygen for a very short time, the back half has probably not suffered any significant damage.
A union can be installed to repair a broken column. Any suitable union will work to rejoin the column. No more than 2-3 unions should be installed on any one column. Problems with dead volume (peak tailing) may occur with multiple unions.
THERMAL DAMAGE:
Exceeding a column upper temperature limit results in accelerated degradation of the stationary phase and tubing surface. This results in the premature onset of excessive column bleed, peak tailing for active compounds and/or loss of efficiency (resolution).
Fortunately, thermal damage is a slower process, thus prolonged times above the temperature limit are required before significant damage occurs. Thermal damage is greatly accelerated in the presence of oxygen. Overheating a column with a leak or high oxygen levels in the carrier gas results in rapid and permanent column damage.
Setting the maximum oven temperature at or a few degrees above the column temperature limit is the best method to prevent thermal damage. This prevents the accidental overheating of the column. If a column is thermally damaged, it may still be functional.
Remove the column from the detector. Heat the column for 8-16 hours at its isothermal temperature limit. Remove 10-15 cm from the detector end of the column. Reinstall the column and condition as usual. The column usually does not return to its original performance; however, it is often still functional. The life of the column will be reduced after thermal damage.
OXYGEN DAMAGE:
Oxygen is an enemy to most capillary GC columns. While no column damage occurs at or near ambient temperatures, severe damage occurs as the column temperature increases.
In general, the temperature and oxygen concentration at which significant damages occurs is lower for polar stationary phases. It is constant exposure to oxygen that is the problem. Momentary exposure such as an injection of air or a very short duration septum nut removal is not a problem.
A leak in the carrier gas flow path (e.g., gas lines, fittings, injector) is the most common source of oxygen exposure. As the column is heated, very rapid degradation of the stationary phase occurs. This results in the premature onset of excessive column bleed, peak tailing for active compounds and/or loss of efficiency (resolution).
These are the same symptoms as for thermal damage. Unfortunately, by the time oxygen damage is discovered, significant column damage has already occurred. In less severe cases, the column may still be functional but at a reduced performance level. In more severe cases, the column is irreversibly damaged.
Maintaining an oxygen and leak free system is the best prevention against oxygen damage. Good GC system maintenance includes periodic leak checks of the gas lines and regulators, regular septa changes, using high quality carrier gases, installing and changing oxygen traps, and changing gas cylinders before they are completely empty.
CHEMICAL DAMAGE:
There are relatively few compounds that damage stationary phases. Introducing non-volatile compounds (high molecular weight or high boiling point) in a column often degrades performance, but damage to the stationary phase does not occur. These residues can often be removed and performance returned by solvent rinsing the column
Inorganic or mineral bases and acids are the primary compounds to avoid introducing in a column. The acids include hydrochloric (HCl), sulfuric (H2SO4), nitric (HNO3), phosphoric (H3PO4) and chromic (CrO3). The bases include potassium hydroxide (KOH), sodium hydroxide (NaOH) and ammonium hydroxide (NH4OH).
Most of these acids and bases are not very volatile and accumulate at the front of the column. If allowed to remain, the acids or bases damage the stationary phase. This results in the premature onset of excessive column bleed, peak tailing for active compounds and/or loss of efficiency (resolution). The symptoms are very similar to thermal and oxygen damage.
Hydrochloric acid and ammonium hydroxide are the least harmful of the group. Both tend to follow any water that is present in the sample. If the water is not or only poorly retained by the column, the residence time of HCl and NH4OH in the column is short. This tends to eliminate or minimize any damage by these compounds. Thus, if HCl or NH4OH are present in a sample, using conditions or a column with no water retention will render these compounds relatively harmless to the column.
The only organic compounds that have been reported to damage stationary phases are perfluoroacids. Examples include trifluoroacetic, pentafluoropropanoic and heptafluorobutyric acid. They need to be present at high levels (e.g., 1% or higher). Most of the problems are experienced with splitless or Megabore direct injections where large volumes of the sample are deposited at the front of the column.
Since chemical damage is usually limited to the front of the column, trimming or cutting 1/2-1 meter from the front of the column often eliminates any chromatographic problems. In more severe cases, 5 or more meters may need to be removed. The use of a guard column or retention gap will minimize the amount of column damage; however, frequent trimming of the guard column may be necessary. The acid or base often damages the surface of the deactivated fused silica tubing which leads to peak shape problems for active compounds.
COLUMN CONTAMINATION
Column contamination is one of the most common problems encountered in capillary GC. Unfortunately, it mimics a very wide variety of problems and is often misdiagnosed as another problem. A contaminated column is usually not damaged, but it may be rendered unusable.
There are two basic types of contaminants: nonvolatile and semi-volatile. Nonvolatile contaminants or residues do not elute and accumulate in the column. The column becomes coated with these residues which interfere with the proper partitioning of solutes in and out of the stationary phase.
Also, the residues may interact with active solutes resulting in peak adsorption problems (evident as peak tailing or loss of peak size). Active solutes are those containing a hydroxyl (-OH) or amine (-NH) group, and some thiols (-SH) and aldehydes.
Semivolatile contaminants or residues accumulate in the column, but eventually elute. Hours to days may elapse before they completely leave the column. Like nonvolatile residues, they may cause peak shape and size problems and, in addition, are usually responsible for many baseline problems (instability, wander, drift, ghost peaks, etc.).
Contaminants originate from a number of sources with injected samples being the most common. Extracted samples are among the worse types. Biological fluids and tissues, soils, waste and ground water, and similar types of matrices contain high amounts of semivolatile and nonvolatile materials.
Even with careful and thorough extraction procedures, small amounts of these materials are present in the injected sample. Several to hundreds of injections may be necessary before the accumulated residues cause problems. Injection techniques such as on-column, splitless and Megabore direct place a large amount of sample into the column, thus column contamination is more common wirh these injection techniques.
Occasionally contaminants originate from materials in gas lines and traps, ferrule and septa particles, or anything coming in contact with the sample (vials, solvents, syringes, pipettes, etc.). These types of contaminants are probably responsible when a contamination problem suddenly develops and similar samples in previous months or years did not cause any problems.
Minimizing the amount of semivolatiles and nonvolatile sample residues is the best method to reduce contamination problems. Unfortunately, the presence and identity of potential contaminants are often unknown. Rigorous and thorough sample cleanup is the best protection against contamination problems. The use of a guard column or retention gap often reduces the severity or delays the onset of column contamination induced problems.
If a column becomes contaminated, it is best to solvent rinse the column to remove the contaminants.
Maintaining a contaminated column at high temperatures for long periods of time (often called baking out a column) is not recommended. Baking out a column may convert some of the contaminating residues into insoluble materials that cannot be solvent rinsed from the column. If this occurs, the column cannot be salvaged in most cases.
Sometimes the column can be cut in half and the back half may still be useable. Baking out a column should be limited to 1-2 hours at the isothermal temperature limit of the column.